

Um grupo interdisciplinar de pesquisadores da Universidade Federal do Ceará, da Universidade de Rice (EUA), de Houston (EUA) e de Edimburgo (Reino Unido) desenvolveu uma plataforma on-line que ajuda cientistas de todo o mundo a testar moléculas com potencial inibitório contra o SARS-CoV-2, o vírus que provoca a covid-19.
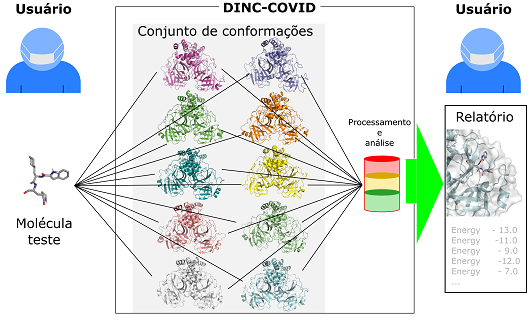
A plataforma, batizada de DINC-COVID, é gratuita e acessível a qualquer cientista. Para utilizá-la, basta o pesquisador fornecer o arquivo tridimensional da molécula que pretende testar, que o sistema realizará as simulações e indicará se há potencial de ligação entre as moléculas testadas e o vírus.
A grande vantagem da ferramenta em relação às técnicas convencionais de ancoramento molecular é que ela leva em consideração a flexibilidade natural das proteínas do SARS-CoV-2 ao testar candidatos a medicamento contra o vírus. Isso aumenta a precisão dos resultados e permite acelerar o processo de identificação e desenvolvimento de novos medicamentos.
COMO É A TÉCNICA
A DINC-COVID usa uma técnica chamada de ancoramento molecular (também conhecida por seu nome em inglês, docking), uma simulação em computador que utiliza as estruturas tridimensionais (3-D) para testar possíveis ligações entre moléculas de determinado organismo (um vírus como o da covid-19, por exemplo) e moléculas candidatas a medicamento.
Para a indústria farmacêutica, essa técnica tem permitido enorme economia de tempo e de recursos: em vez de realizar milhares de testes de bancada em busca de uma molécula que faça determinada ação, apenas aquelas que conseguirem interagir com o organismo-alvo na simulação passam para as etapas seguintes da pesquisa.
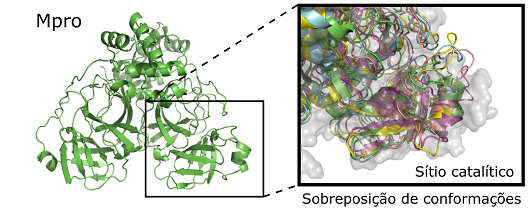
Nas suas origens, os sistemas de ancoramento molecular rodavam simulações considerando as imagens estáticas de proteínas, moléculas e vírus. Era um modelo simples de “chave-fechadura”, longe do que ocorre realmente no corpo humano, onde essas estruturas são flexíveis e estão em movimento. Versões mais recentes da técnica passaram a considerar o movimento parcial das moléculas estudadas, contudo essas abordagens demandam alto poder computacional e o emprego simultâneo de várias técnicas de simulação.
COMO NASCEU A IDEIA
No começo da pandemia, o Prof. Geancarlo Zanatta, do Departamento de Física da UFC, acompanhou com atenção o enorme esforço ‒ e a falta de sucesso ‒ de inúmeros grupos de pesquisa em busca de moléculas que pudessem ser utilizadas no bloqueio do vírus. O tema interessava particularmente a Zanatta, farmacêutico de formação, com estudos na área de biologia estrutural e experiência em ancoramento molecular.
A dificuldade, lembra ele, é que nem todos os grupos dispunham dos recursos mais atualizados para a realização do docking. “Muitos pesquisadores possuíam moléculas para testar, mas por trabalharem em outras áreas desconheciam aspectos técnicos importantes para essa abordagem”, diz. Para ser mais efetivo, seria necessário que eles adotassem justamente um instrumento que considerasse a flexibilidade das proteínas no corpo humano.
Dessa constatação, surgiu a ideia de uma ferramenta aberta que pudesse ser usada em rede, ajudando o trabalho de outros grupos. A iniciativa do Prof. Zanatta foi abraçada pelo grupo coordenado pela Profª Lydia Kavraki, da área de ciência da computação, na Universidade de Rice, e pelo pesquisador brasileiro Dinler Antunes, que realizava pós-doutorado em Rice e atualmente é professor na Universidade de Houston.
O trabalho foi encabeçado pelos pesquisadores Sarah Hall-Swan, da própria Rice, e Didier Devaurs, da Universidade de Edimburgo. Ao grupo, se juntou Mauricio Rigo, que realiza pós-doutorado na Rice.
A equipe extraiu grandes conjuntos de informações do Protein Data Bank, um enorme banco de dados de moléculas biológicas em 3-D, alimentado por cientistas de todo o mundo. Depois, incluiu dados de inúmeras simulações de dinâmica molecular rodadas com o auxílio de supercomputadores brasileiros.

O grupo passou um ano processando informações e desenvolvendo a plataforma on-line. Para trazer mais confiabilidade, a equipe utilizou-se do código DINC, ferramenta de ancoramento molecular desenvolvida pela equipe americana e consolidada entre a comunidade científica.
Atualmente, estão disponibilizados 15 conjuntos de conformações de proteínas virais, representando sete regiões no vírus da covid-19 que podem ser inibidas. Quando um pesquisador quer testar uma molécula, é nesses conjuntos que as simulações são aplicadas.
O resultado do trabalho foi disponibilizado no início deste ano em formato preprint (preliminar, sem a revisão de pares) e recentemente publicado na revista Computers in Biology and Medicine. Ainda no início de 2021, o servidor foi disponibilizado.
De acordo com o Prof. Zanatta, desde então mais de 500 cientistas de 53 países já utilizaram a DINC-COVID em suas pesquisas. Com a publicação da versão definitiva, a expectativa é que a quantidade de usuários cresça.
Fonte: Prof. Geancarlo Zanatta, do Departamento de Física da UFC ‒ e-mail: geancarlo.zanatta@fisica.ufc.br


